I've run into an "interesting" issue with DNSWL (a great whitelisting/blacklisting service) recently. As part of their DNS query abuse/freeloading mitigation, DNSWL blocks DNS queries (or returns results that marks all hosts as high-confidence non-spam sources) for hosts which repeatedly exceed reasonable query limits to help funnel large users to their subscription service.
Requests from my mail host to dnswl via IPv6 started to trigger the more serious mitigation where DNSWL returns bogus results (every result is high confidence that it is not spam).
It appears that they are binning requests via IPv6 into large blocks (/64 or larger). As my mail host runs in a larger hosting provider's network, all of the DNS requests in that block are binned together, and exceed the limits, resulting in bogus results.
Bind has an interesting feature where you can mark certain DNS servers as "bogus" to ignore query results from them.. Using this feature, we can ignore the dnswl IPv6 servers which are returning bad results, and only use IPv4 to contact them:
server 2a01:7e00:e000:293::a:1000 { bogus yes; };
server 2607:5300:201:3100::3e79 { bogus yes; };
server 2600:3c01::21:1faa { bogus yes; };
server 2a01:4f8:c2c:52e::feed { bogus yes; };
server 2400:8901::f03c:91ff:fee9:a89 { bogus yes; };
server 2a01:4f8:1c0c:708f::53 { bogus yes; };
Hope that helps anyone else (and future me) running into this issue.
One of the key features of Debbugs, the bug tracking system Debian uses, is its ability to figure out which bugs apply to which versions of a package by tracking package uploads. This system generally works well, but when a package maintainer's workflow doesn't match the assumptions of Debbugs, unexpected things can happen. In this post, I'm going to:
- introduce how Debbugs tracks versions
- provide an example of a merge-based workflow which Debbugs doesn't handle well
- provide some suggestions on what to do in this case
Debbugs Versioning
Debbugs tracks versions using a set of one or more rooted trees which it builds from the ordering of debian/changelog entries. In the simplist case, every upload of a Debian package has changelogs in the same order, and each upload adds just one version. For example, in the case of dgit, to start with the package has this (abridged) version tree:

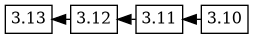
the next upload, 3.13, has a changelog with this version ordering:
3.13 3.12 3.11 3.10, which causes the 3.13 version to be added as
a descendant of 3.12, and the version tree now looks like this:
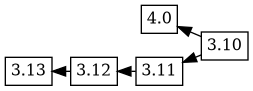
dgit is being developed while also being used, so new versions with
potentially disruptive changes are uploaded to experimental while
production versions are uploaded to unstable. For example, the 4.0
experimental upload was based on the 3.10 version, with the
changelog ordering 4.0 3.10. The tree now has two branches, but
everything seems as you would expect:
Merge based workflows
Bugfixes in the maintenance version of dgit also are made to the experimental package by merging changes from the production version using git. In this case, some changes which were present in the 3.12 and 3.11 versions are merged using git, corresponds to a git merge flow like this:
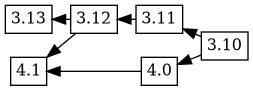
If an upload is prepared with changelog ordering 4.1 4.0 3.12 3.11
3.10, Debbugs combines this new changelog ordering with the
previously known tree, to produce this version tree:
This looks a bit odd; what happened? Debbugs walks through the new
changelog, connecting each of the new versions to the previous version
if and only if that version is not already an ancestor of the new
version. Because the changelog says that 3.12 is the ancestor of
4.0, that's where the 4.1 4.0 version tree is connected.
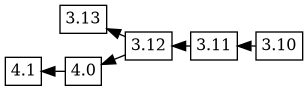
Now, when 4.2 is uploaded, it has the changelog ordering (based on
time) 4.2 3.13 4.1 4.0 3.12 3.11 3.10, which corresponds to this git
merge flow:
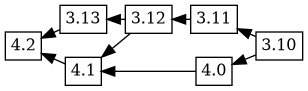
Debbugs adds in 3.13 as an ancestor of 4.2, and because 4.1 was not an ancestor of 3.13 in the previous tree, 4.1 is added as an ancestor of 3.13. This results in the following graph:

Which doesn't seem particularly helpful, because
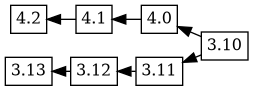
is probably the tree that more closely resembles reality.
Suggestions on what to do
Why does this even matter? Bugs which are found in 3.11, and fixed in 3.12 now show up as being found in 4.0 after the 4.1 release, though they weren't found in 4.0 before that release. It also means that 3.13 now shows up as having all of the bugs fixed in 4.2, which might not be what is meant.
To avoid this, my suggestion is to order the entries in changelogs in
the same order that the version graph should be traversed from the
leaf version you are releasing to the root. So if the previous version
tree is what is wanted, 3.13 should have a changelog with ordering
3.13 3.12 3.11 3.10, and 4.2 should have a changelog with ordering
4.2 4.1 4.0 3.10.
What about making the BTS support DAGs which are not trees? I think something like this would be useful, but I don't personally have a good idea on how this could be specified using the changelog or how bug fixed/found/absent should be propagated in the DAG. If you have better ideas, email me!
Like many laptop users, I often plug my laptop into different monitor setups (multiple monitors at my desk, projector when presenting, etc.) Running xrandr commands or clicking through interfaces gets tedious, and writing scripts isn't much better.
Recently, I ran across autorandr, which detects attached monitors using EDID (and other settings), saves xrandr configurations, and restores them. It can also run arbitrary scripts when a particular configuration is loaded. I've packed it, and it is currently waiting in NEW. If you can't wait, the deb is here and the git repo is here.
To use it, simply install the package, and create your initial configuration (in my case, undocked):
autorandr --save undocked
then, dock your laptop (or plug in your external monitor(s)), change the configuration using xrandr (or whatever you use), and save your new configuration (in my case, workstation):
autorandr --save workstation
repeat for any additional configurations you have (or as you find new configurations).
Autorandr has udev, systemd, and pm-utils hooks, and autorandr
--change should be run any time that new displays appear. You can
also run autorandr --change or autorandr --load workstation
manually too if you need to. You can also add your own
~/.config/autorandr/$PROFILE/postswitch script to run after a
configuration is loaded. Since I run i3, my workstation configuration
looks like this:
#!/bin/bash
xrandr --dpi 92
xrandr --output DP2-2 --primary
i3-msg '[workspace="^(1|4|6)"] move workspace to output DP2-2;'
i3-msg '[workspace="^(2|5|9)"] move workspace to output DP2-3;'
i3-msg '[workspace="^(3|8)"] move workspace to output DP2-1;'
which fixes the dpi appropriately, sets the primary screen (possibly
not needed?), and moves the i3 workspaces about. You can also arrange
for configurations to never be run by adding a block hook in the
profile directory.
Check it out if you change your monitor configuration regularly!


One of our cats (Haru) chews small wires, and recently chewed through the USB Type A to barrel connector cord for my LED reading light. No biggie, I thought, I'll just buy a replacement for it, and move on. But wait, an entirely new reading light is just as cheap! I'll buy that so I'll have two, and I won't have to worry about buying the wrong connector.
When the new reading light arrived, I found out it now used a micro USB connector instead of the barrel, and more importantly, wouldn't run off of the battery. Some disassembly, and the reason became pretty obvious. The battery was slightly bulgy, had almost no resistance, and had zero voltage. All signs of a battery which shorted out at some point very early in its lifetime.
Luckily, I had a working battery from the older light... so why not swap? Some dodgy soldering work later, and voila! One working light with more universal connectors and some extra parts. My rudimentary soldering and electronic troubleshooting skills keep coming in handy.
Our paper which describes the components of the placenta transcriptome which are conserved among all placental mammals in Placenta just came out today. More importantly than the results and the text of the paper, though, is the fact that all of the code and results of this paper, from the very first work I did two years ago to its publication today is present in git, and (in theory) reproducible.
You can see where our paper was rejected from Genome Biology and Genes and development and radically refocused before submission to Placenta. But more importantly, you can know where every single result which is mentioned in the paper came from, the precise code to generate it, and how we came to the final paper which was published. [And you've also got all of the hooks to branch off from our analysis to do your own analysis based on our data!]
This is what open, reproducible science should look like.
I've been trying to finish a paper where I compare gene expression in 14 different placentas. One of the supplemental figures compares median expression in gene trees across all 14 species, but because tree ids like ENSGT00840000129673 aren't very expressive, and names like "COL11A2, COL5A3, COL4A1, COL1A1, COL2A1, COL1A2, COL4A6, COL4A5, COL7A1, COL27A1, COL11A1, COL4A4, COL4A3, COL3A1, COL4A2, COL5A2, COL5A1, COL24A1" take up too much space, I wanted a function which could collapse the gene names into something which uses bash glob syntax to more succinctly list the gene names, like: COL{11A{1,2},1A{1,2},24A1,27A1,2A1,3A1,4A{1,2,3,4,5,6},5A{1,2,3},7A1}.
Thus, a crazy function which uses lcprefix from Biostrings and
some looping was born:
collapse.gene.names <- function(x,min.collapse=2) {
## longest common substring
if (is.null(x) || length(x)==0) {
return(as.character(NA))
}
x <- sort(unique(x))
str_collapse <- function(y,len) {
if (len == 1 || length(y) < 2) {
return(y)
}
y.tree <-
gsub(paste0("^(.{",len,"}).*$"),"\\1",y[1])
y.rem <-
gsub(paste0("^.{",len,"}"),"",y)
y.rem.prefix <-
sum(combn(y.rem,2,function(x){Biostrings::lcprefix(x[1],x[2])}) >= 2)
if (length(y.rem) > 3 &&
y.rem.prefix >= 2
) {
y.rem <-
collapse.gene.names(y.rem,min.collapse=1)
}
paste0(y.tree,
"{",paste(collapse=",",
y.rem),"}")
}
i <- 1
ret <- NULL
while (i <= length(x)) {
col.pmin <-
pmin(sapply(x,Biostrings::lcprefix,x[i]))
collapseable <-
which(col.pmin > min.collapse)
if (length(collapseable) == 0) {
ret <- c(ret,x[i])
i <- i+1
} else {
ret <- c(ret,
str_collapse(x[collapseable],
min(col.pmin[collapseable]))
)
i <- max(collapseable)+1
}
}
return(paste0(collapse=",",ret))
}
I'm in Pretoria, South Africa at the H3ABioNet hackathon which is developing workflows for Illumina chip genotyping, imputation, 16S rRNA sequencing, and population structure/association testing. Currently, I'm working with the imputation stream and we're using Nextflow to deploy an IMPUTE-based imputation workflow with Docker and NCSA's openstack-based cloud (Nebula) underneath.
The OpenStack command line clients (nova and cinder) seem to be
pretty usable to
automate bringing up a fleet of VMs
and the cloud-init package which is present in the images makes
configuring the images pretty simple.
Now if I just knew of a better shared object store which was supported by Nextflow in OpenStack besides mounting an NFS share, things would be better.
You can follow our progress in our git repo: [https://github.com/h3abionet/chipimputation]
Many bioinformatic problems require large amounts of memory and processor time to complete. For example, running WGCNA across 10⁶ CpG sites requires 10⁶ choose 2 or 10¹³ comparisons, which needs 10 TB to store the resulting matrix. While embarrassingly parallel, the dataset upon which the regressions are calculated is very large, and cannot fit into main memory of most existing supercomputers, which are often tuned for small-data fast-interconnect problems.
Another problem which I am interested in is computing ancestral trees from whole human genomes. This involves running maximum likelihood calculations across 10⁹ bases and thousands of samples. The matrix itself could potentially take 1 TB, and calculating the likelihood across that many positions is computationally expensive. Furthermore, an exhaustive search of trees for 2000 individuals requires 2000!! comparisons, or 10²⁸⁶⁸; even searching a small fraction of that subspace requires lots of computational time.
Some things that a future supercomputer could have that would enable better solutions to bioinformatic problems include:
- Fast local storage
- Better hierarchical storage with smarter caching. Data should ideally move easily between local memory, shared memory, local storage, and remote storage.
- Fault-tolerant, storage affinity aware schedulers.
- GPUs and/or other coprocessors with larger memory and faster memory interconnects.
- Larger memory (at least on some nodes)
- Support for docker (or similar) images.
- Better bioinformatics software which can actually take advantage of advances in computer architecture.
Having a new student join me to work in the lab reminded me that I should collect some of the many resources around for getting started in bioinformatics and any data-based science in general. So towards this end, one of the first essential tools for any data scientist is a knowledge of git.
Start first with Code School's simple introduction to git which gives you the basics of using git from the command line.
Then, check out set of lectures on Git and GitHub which goes into setting up git and using it with github. This is a set of lectures which was used in a Data Science course.
Finally, I'd check out the set of resources on github for even more information, and then learn to love the git manpages.
I've been using qsub for a while now on the cluster here at the IGB at UofI. qsub is a command line program which is used to submit jobs to a scheduler to eventually be run on one (or more) nodes of a cluster.
Unfortunately, qsub's interface is horrible. It requires that you write a shell script for every single little thing you run, and doesn't do simple things like providing defaults or running multiple jobs at once with slightly different arguments. I've dealt with this for a while using some rudimentary shell scripting, but I finally had enough.
So instead, I wrote a wrapper around qsub called dqsub.
What used to require a complicated invocation like:
echo -e '#!/bin/bash\nmake foo'| \
qsub -q default -S /bin/bash -d $(pwd) \
-l mem=8G,nodes=1:ppn=4 -;
can now be run with
dqsub --mem 8G --ppn 4 make foo;
Want to run some command in every single directory which starts with SRX? That's easy:
ls -1 SRX*|dqsub --mem 8G --ppn 4 --array chdir make bar;
Want instead to behave like xargs but do the same thing?
ls -1 SRX*|dqsub --mem 8G --ppn 4 --array xargs make bar -C;
Now, this wrapper isn't complete yet, but it's already more than enough to do what I require, and has saved me quite a bit of time already.
You can steal dqsub for yourself
Feel free to request specific features, too.
This blog is powered by ikiwiki.